Pfeifferin oireyhtymä: syyt, oireet, hoito
Pfayfferin oireyhtymä on erittäin harvinaistageneettinen sairaus, joka esiintyy keskimäärin yhdestä 100 000 vastasyntyneestä. Vaikuttaa yhtä usein pojille ja tytöille. Tärkein taudin oire on alkukahvaisten luuston luuston varhainen yhteensumi- nen alkion alkamisprosessissa, jonka vuoksi aivot eivät enää voi kehittyä normaalisti.
Yleistietoja
Tauti löysi kuuluisa saksalainengenetisti Rudolf Pfeiffer. Vuonna 1964 hän kuvaili aikaisemmin tuntemattoman sairauden oireita, joiden pääasialliset merkit olivat epänormaalit kallon rakenteessa ja sormien liittäminen jalkoihin tai käsiin (syndaattisesti). Oireyhtymää havaittiin kahdeksassa samaan perheeseen kuuluvasta potilaasta, jotka osoittivat taudin geneettisen luonteen. Muita tutkimuksia ovat osoittaneet, että Pfeifferin oireyhtymä on siirretty autosomaalisella dominantilla.
Ultrasound voi havaita tämän sairauden sikiössä jo toisen raskauskolmanneksen aikana. Ultrasuurilla nähdään kallon, sisäelinten ja ääripäiden kehityksen epänormaaleja ominaisuuksia.
oireet
Vuonna 1993 amerikkalainen geneetikko Michael CohenHän ehdotti Pfeifferin oireyhtymän luokittelua kolmeen tyyppiin riippuen oireiden vakavuudesta. Nyt luokittelu on yleisesti hyväksytty ja sitä käytetään laajalti lääketieteellisessä kirjallisuudessa.
Pfayffer-oireyhtymän tyypin 1 oireita ovat mmKraniosynostosi - ehto, jossa yksi tai useampi kraniaalinen sutura yhdistyy ennenaikaisesti muodostaen kokonaisvaltaisen luun. Tuloksena on havaittavissa muutoksia kallon muotoon ja epänormaaleihin kasvojen piirteisiin - hypertelorismi, alhaiset korvat. Voi olla heikentynyt, kohonnut kallonsisäinen paine, oikosulake. 50%: lla potilaista on havaittu kuulon heikkenemistä. Useimmilla tällaisen sairauden omaavilla ihmisillä on normaali älykkyys ja heillä ei ole neurologisia poikkeavuuksia.
Lapsilla, joilla on tyypin 1 Pfayffer-oireyhtymä, ulkoinenMerkkejä, kuten laaja lyhyt sormi käsivarret ja jalat, havaitaan lähes aina. Syndykytys on mahdollinen, mutta ei välttämätön oire tämän tyyppiselle patologialle.

Tämäntyyppinen oireyhtymä on periytynyt autosomaalisesti hallitsevasti.
Alla on kuva Pfeifferin oireyhtymästä. Elinajanodote Venäjällä tämän taudin kärsiville ihmisille ylittää 60 vuotta.

Tyypin 2 sairaudesta kallolle on tunnusomaista akeisariksen muoto, selkärangan fuusio, syndykytys ja silmämunien siirtyminen eteenpäin (proptosis). Elämän ensimmäisistä päivistä ilmenee vakavia neurologisia häiriöitä.
Kolmantyyppistä tautia on tyypillistä vieläkin enemmänvarhainen kraniosynostosis. Kallo on epänormaalisti pitkänomainen ja ei muodosta "kalkkuna". Hampaiden poikkeavuuksia, hypertelorismia, henkistä hidastumista ja sisäelinten epämuodostumia esiintyy.
2. ja 3. tyypin elinikätauti vaihtelee useista päivistä useisiin kuukausiin - sisäelinten puutteet ja neurologiset häiriöt eivät ole yhteensopivia elämän kanssa. Näistä kahdesta Pfeiffer-oireyhtymästä vastaavat mutaatiot esiintyvät satunnaisesti eikä geneettisesti periytyviä.
syistä
Pfayfferin oireyhtymä liittyy reseptorin 1 mutaatioihinfibroblastikasvutekijä (FGFR1) kromosomista 8 tai fibroblastikasvutekijäreseptori 2 (FGFR2) kromosomissa 10. geenejä, jotka vaikuttavat vastuussa olevan mutaation normaalin kasvun fibroblastien, jolla on keskeinen rooli kehon luun kehittymiseen.

hoito
Nykyaikainen lääke ei vielä ole mahdollista poistaamutaatioita ihmisen genomissa, vaikka tiedetään, missä geeneissä on ollut perestroika. Siksi Pfeiffer-oireyhtymälle, kuten useimmille muille geneettisille sairauksille, hoito on oireileva. Tyypin 2 ja tyypin 3 potilailla ennuste on epäsuotuisa: lukuisia kehityshäiriöitä ei voida oikaista lääketieteellisesti tai kirurgisesti.
Pfayfferin tyypin 1 oireyhtymä on yhteensopiva elämän kanssa. Tärkein sairauden oire - kallon luiden ennenaikainen fuusio - voidaan korjata kirurgisesti. Toimenpide suositellaan lapsille aina kolmen kuukauden ikäiseksi.
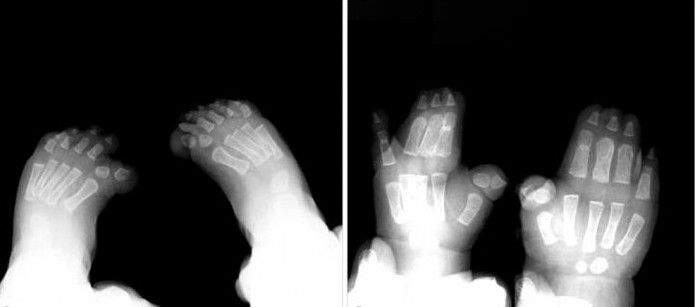
Lisäksi kirurginen interventio on tehokas yksinkertaisen syndaktyylin tapauksessa. Hoito on parasta aloittaa 18-24 kuukauden iässä, sormien ja varpaiden aktiivisen kasvun aikana.







